2.3. Basic energy science
2.3.1. Photochemistry
Understanding and controlling photochemical reactions is the key to mastering some of the most important processes to be addressed in energy and environmental science such as artificial photosynthesis and water splitting, and CO2 recycling. Understanding how the correlated motion of electrons and nuclei is linked to the reaction kinetics and controlling their coupled dynamics clearly motivates the need for information on photoexcited electronic and structural changes in molecular systems and in particular on their temporal evolution. The key is to capture intermediate molecular states that may occur during photochemical reactions and to reveal their sequence through transient configurations all the way from educt to product states. This is the basis for understanding photochemical reactions as it allows for elucidating the elementary reaction steps of molecular dissociation, charge transfer and isomerisation. Furthermore, it is the basis for learning how to control photochemical reactions, i.e., to learning how to drive reactions along desired pathways and how to drive them to desired products.
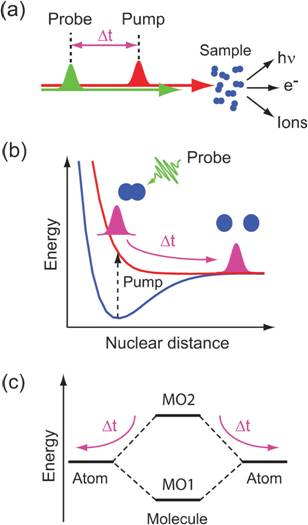
Figure 15. Schematic representation of (a) a pumpprobe experiment, (b) the nuclear dynamics of a molecule in a dissociation reaction of a diatomic molecule and (c) a molecular orbital level scheme illustrating the transient electronic structure in a dissociation reaction of a diatomic molecule. (b) The nuclear dynamics of the photo-excited and dissociating molecules are represented with nuclear wavepackets (magenta) in a potential energy diagram. The pump pulse induces a transition from the ground state of the molecule (blue) to a dissociative state (red) and the evolution of the nuclear wavepacket is probed with the probe pulses (green) at time delays Δt after initiation of the dissociation. (c) The electronic structure of the diatomic molecule is represented in an orbital energy level diagram with two arbitrary molecular orbitals MO1 and MO2 in the molecule at bonding distance of the nuclei and atomic orbitals for infinite nuclear distances. Taken from ref. 1, Wernet, PCCP 13, 16941 (2011).
Understanding photochemical processes requires following the structural dynamics and the valence charge density ground and excited transient states (Figure 15). Especially the latter can be uniquely probed via time-resolved X-ray spectroscopy because it provides a complete picture of chemical bonding from the perspective of selected atomic sites. This applies in particular to third-row transition-metal complexes which play a key role in most areas of chemistry and are vital for essentially all forms of life. The chemistry of the transition-metal complexes is largely determined by the metal-3d valence charge density. Dipole-allowed core-level excitations of the metal-2p electrons provide an excellent probe thereof, revealing symmetry, delocalization, and spin-state of ground and transient states (Figure 16).
These transitions lie in the soft X-ray range covered by BESSY-VSR, where lighter elements that are essential in photochemical processes as well, carbon, nitrogen, and oxygen, can be uniquely probed via 1s core-level excitations. These elements typically act as the coordinating nearest-neighbor atoms of the metal center and probing their core-level transitions delivers the complementary ligand view. Furthermore, probes of non-metallic chemistry can be accessed, allowing for the study of elementary photochemistry in organic molecules.
X-ray spectroscopy at BESSY-VSR will uniquely allow for investigating the evolution of the valence charge distribution and bonding during ultrafast photochemical dynamics. The increased flux at the femtoslicing facility FEMTOSPEX at BESSY-VSR will allow for femtosecond X-ray absorption spectroscopy for mapping the transient changes in the valence-charge distribution during ultrafast molecular transformation of dilute solvated species.
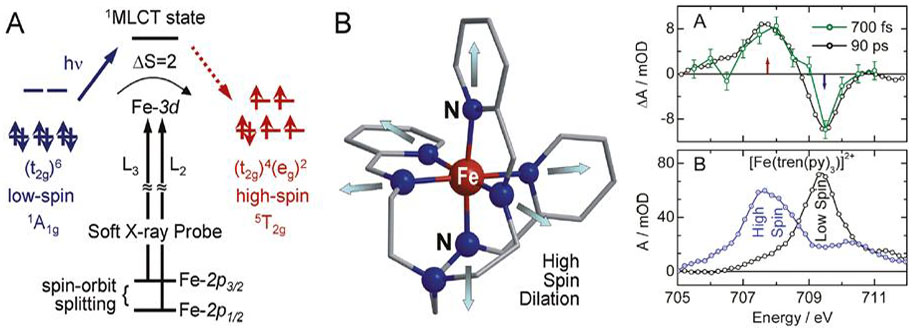
Figure 16. Left: (A) Qualitative orbital diagram for an octahedral Fe(II) complex, illustrating how the photoinitiated spin crossover is probed via metal 2pà3d dipole transitions. (B) Schematic drawing of the molecular structure of [Fe(tren(py)3)]2+. Photoinduced metal-to-ligand charge transfer results in ultrafast structural changes that have been previously characterized by picosecond time-resolved EXAFS measurements. Right: (A) Time-resolved differential X-ray absorption spectra of [Fe(tren(py)3)]2+ in CH3CN solution at the Fe L3-edge. (B) L3-edge spectra of [Fe(tren(py)3)]2+ in CH3CN in the low-spin ground state (black) and the transient high-spin excited state (blue) at Δt = 90 ps. Taken from ref. 2, Huse et al., J. Phys. Chem. Lett. 2, 880 (2011).
X-ray spectroscopy with the few-ps pulses at BESSY-VSR will in addition allow for addressing the subsequent and more subtle changes of chemical bonding resulting from solvation, complexation and subtle but essential changes of molecular symmetry in photochemical reaction intermediates. With this, BESSY-VSR will uniquely complement studies on the femtosecond time scale from free-electron lasers on the one hand and investigations on the ns time scale at current synchrotrons.
Charge-transfer reactions in metalloenzymes
Metalloenzymes are present in all areas of cellular metabolism and display remarkable properties through coupling of transition metal complexes to the amino acids of a proteins peptide chain. Metalloenzymes are thus capable of catalyzing (multi)electron chemical reactions while circumventing highly reactive radical intermediates or oxidizing unwanted radical side products of other metabolic cell activity, thereby reducing the influence of toxic species. The inverse process has also been observed, making use of cell toxicity of certain substances for immune defense or infiltration of host cells. Prominent examples of metalloproteins and -enzymes are myoglobin (Figure 17) the oxygen-binding protein of muscle tissue in vertebrates and the photosystems of plants in the reaction center of which a manganese complex splits water subsequent to absorption of sunlight, leading to the well-known photosynthesis (section 2.4.1). Generally, metalloenzymes use many oxidation states of transition metal complexes (mainly vanadium to zinc) for their catalytic function. The metal reaction centers gain specific activity by lowered potential barriers and undergo redox cycles during substance turnover.
The role of metal centers during chemical reactions or phase transitions can be uniquely studied via X-ray spectroscopy due to their characteristic absorption in the X-ray regime. The corresponding core-level transitions are not only element-specific but deliver information of oxidation and spin states as well as changes in metal-ligand interactions which determine the chemistry of the valence charge density.
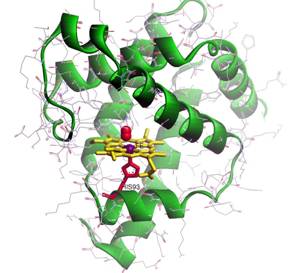
Figure 17. Structural model of Myoglobin emphasizing the heme cofactor. Atoms shown in purple (red) correspond to Fe (O) (adapted from http://www.fizyka.umk.pl/~wiesiek/).
The combination of results from time-resolved X-ray spectroscopic studies from BESSY-VSR and state-of-the-art simulations will allow gaining unique insight into reaction mechanisms and their intermediates on fs and ps time scales. Access to information on valence charge density is complementary to structural methods such as diffraction and scattering, delivering essential ingredients for a comprehensive understanding of transient states and chemical transformations of matter. Possible reaction triggers have traditionally been photoinduced charge-transfer reactions but additional chemical triggers such as heat and pH jumps as well as use of caged compounds will allow for the study of any other classes of chemical reactions.
Photo-triggered proton-transfer reactions in solution
Proton transfer is a key process in nature and can be considered as important as electron transfer. Essential functionality of biological systems in terms of cellular energy management is accomplished by proton transfer mechanisms which are provided by proton pump proteins. Proton transfer is a key mechanism behind the hydrogen bond, e.g. in DNA, and might lead to disturbance of the genetic code through formation of tautomers of the base pairs (Figure 18). The proton transport mechanism at the intermolecular level, e.g. in aqueous solutions is currently described by a proton hopping mechanism through water bridges [3, 4]. On the intramolecular level, proton transport occurs by means of proton donor and proton acceptor sites within complex molecules. In order to understand how protons transfer, an element-specific look at the molecular groups involved in the proton transfer process is highly desirable.
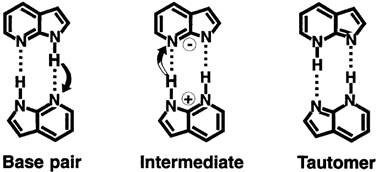
Figure 18. Molecular structures involved in the cooperative double-proton transfer dynamics of the base pair 7-azaindole dimer. Taken from ref. 5, Douhal et al., Nature 378, 260 (1995).
The unique possibility offered at BESSY-VSR with time-resolved X-ray absorption spectroscopy is in the investigation of photoinduced proton transfer. In a femtosecond pump-probe setup the proton transfer will be initiated by an optical pump pulse. The changes in the electronic and nuclear wavefunction of the molecule at the selected atomic center will then be probed by transient X-ray absbsorption. By this, the ultrafast dynamics of elementary biological processes become accessible on the femto- to picosecond time scale, i.e. in photo-tautomerization [5, 6].
Nonequilibrium processes in hydrogen-bonded systems
Hydrogen bonds (HBs) constitute one of the most important interactions in a large class of (bio)molecular systems. Compared with covalent bonds, hydrogen bonds are relatively week but their directional nature is important is essential to a manifold of structures, chemical processes and physical properties. For instance, the special properties of water are largely due to the possibility of water molecules to form tetrahedrally arranged hydrogen bonds, forming a rapidly fluctuating network of dissolving and reforming bonds. The resulting solvent properties are essential for all living matter and are also relevant for the global climate. Furthermore, secondary structures of proteins (a-helices and b-sheets) are generated by hydrogen bonds among the building blocks of proteins (amino acids). Tertiary contacts between protein subunits are often formed via hydrogen bonds as well. Furthermore, hydrogen bonds are vital for the incorporation and functionality of protein cofactors small molecules such as the oxygen-binding heme group in of the relevant proteins in human blood.
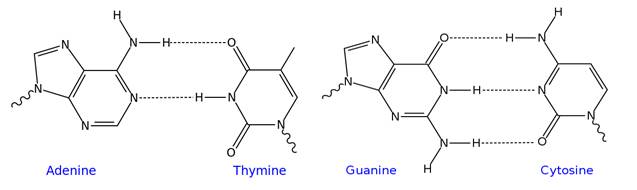
Figure 19. Schematics of the hydrogen-bonded adenine-thymine and guanine-cytosine building blocks of DNA. Adapted from Wikipedia.org, author: Iñaki Silanes.
The multitude of hydrogen bonds formed between stacked base pairs (shown schematically in Figure 19) in combination with interactions of p-electron systems allows for very stable structures (e.g. DNA helices) as well as the possibility of breaking double-stranded helical arrangements to allow for genome replication, essential for cell division (replication and translation) and alterations of genetic mutations avoiding tumors (repair). Time-resolved X-ray spectroscopy can track changes in valence charge density of hydrogen-bonded systems (Figures 20, 21) undergoing chemical transformations by exploiting the spectrally well-separated core-level transitions of light elements with atomic resolution intrinsic to such probe transitions.
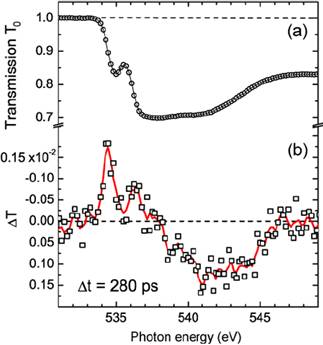
Figure 20. (a) X-ray transmission of liquid water at the O K-edge. (b) Transient changes of the x-ray transmission ÄT = (T − T0) with T0 and T transmission before and after excitation at a pump-probe delay time of 280 ps as a function of photon energy across the O K-edge. Measured raw data are shown as markers (squares), and smoothed data are shown as solid line. Taken from ref. 7, Wernet et al., Appl Phys A 92, 511 (2008).
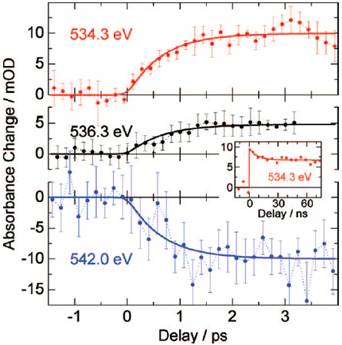
Figure 21. Absorbance change as a function of time delay between the infrared pump pulses and the X-ray probe pulses at three characteristic spectral positions in the O K-edge: 534.3 eV (red), 536.3 eV (black), and 542.0 eV (blue). Solid lines are single exponential fits to the data with a time constant of 0.7 ps. The inset shows a time transients on nanosecond time scales. Taken from ref. 8, Wen et al., J Chem. Phys. 131, 234505 (2009).
BESSY-VSR with the available temporal resolution from fs to ps covers the essential time scales for unprecedented insight into the nonequilibrium processes in hydrogen-bonded systems. Compared to the present situation at BESSY II where hydrogen-bond dynamics were studied water (Figures 20, 21), BESSY-VSR with increased flux for temporal resolutions in the femtosecond to few-ps regime will in particular allow investigating the dynamics of dilute hydrogen-bonded systems such as solvated organic compounds (Figure 22) and nucleic acids with time-resolved X-ray absorption spectroscopy for the first time.
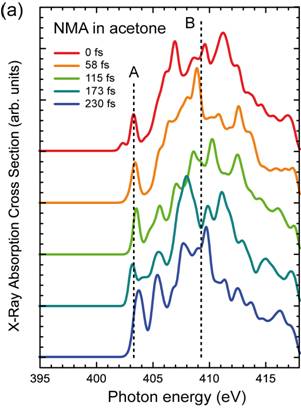
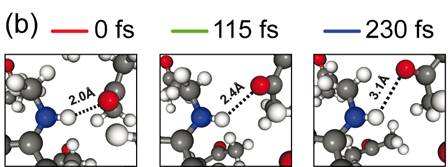
Figure 22. (a) Calculated time evolution of the X-ray absorption spectra at the N K-edge of N-methylaniline in acetone as calculated with configurations derived from an ab initio MD simulations during a hydrogen bond breaking event. The dashed lines mark the resonance maxima of the pre (A) and main (B) edges for the spectrum at 0 fs. (b) Molecular movie depicting zoomed-in views of the N-H···O hydrogen-bond for 0, 115 and 230 fs from (a) as derived from the ab initio MD simulations. Taken from ref. 9, Prémont-Schwarz et al., in preparation (2013).
References for section 2.3.1.
[1] Ph. Wernet, PCCP 13, 16941 (2011).
[2] N. Huse et al., J. Phys. Chem. Lett. 2, 880 (2011).
[3] O. F. Mohammed, et.al., Science 310 , 83 (2005).
[4] N. Agmon, Chemical Physics Letters 244, 456 (1995).
[5] A. Douhal et al., Nature 378, 260 (1995).
[6] O. K. Abou-Zied, V. Tsui, R. Jimenez, D. A. Case, F. E. Romesberg, J. Am. Chem. Soc. 122, 9917 (2000).
[7] Ph. Wernet et al., Appl Phys A 92, 511 (2008).
[8] H. Wen et al., J Chem. Phys. 131, 234505 (2009).
[9] M. Prémont-Schwarz, S. Schreck, M. Iannuzzi, E. T. J. Nibbering, M. Odelius, Ph. Wernet, in preparation (2013).