Abteilung KI und Biomolekulare Strukturen
Forschung
Strukturbiologische Methoden und Fallstudien
Die Bestimmung biomolekularer Strukturen mittels Kristallographie und Kryo-EM ist die Grundlage der modernen Molekularbiologie und Biochemie. Diese Strukturen lassen sich jedoch nicht allein aus experimentellen Daten gewinnen – zur Interpretation der gemessenen Bilder benötigt man ein Modell, und darum ist das Verständnis der zugrundeliegenden Prinzipien von entscheidender Bedeutung. Wenn wir dieses Verständnis noch weiter vertiefen können, können wir mehr Informationen aus unseren experimentellen Daten gewinnen und schwierigere Strukturen lösen.
Wir arbeiten in sehr unterschiedlichen Feldern, von KI-Algorithmen bis hin zur Laborarbeit; der Großteil unserer Forschung wird von praktischen Problemen getrieben und ist oft kooperativ. Allen gemeinsam ist, dass wir mehr über das Aussehen und die Funktion von biologischen Molekülen herausfinden wollen!
Aktuelle Forschungsschwerpunkte
Künstliche Intelligenz in der Strukturbiologie
Wir nutzen KI-Methoden (genauer gesagt maschinelles Lernen), um die experimentelle Strukturbiologie voranzutreiben. Wir evaluieren und verbessern experimentelle Datenmessungen und -verarbeitung im Verbundprojekt AUSPEX, interpretieren 3D-Dichte aus Einpartikel-Kryoelektronenmikroskopie und erforschen, wie man Daten von verschiedenen Methoden und aus der ganzen Welt mit KI kombinieren kann. Bisher umfasste dies die Interpretation von 2D- und 3D-Daten mit neuronalen Netzen und statistischen Methoden sowie Clustering. Wir möchten aber auch GANs zur Simulation experimenteller Daten einsetzen und haben nach dem Ende der Coronavirus Structural Task Force im Februar 2023 begonnen, uns mit Fragen zu linguistischen Modellen in der integrativen Strukturbiologie zu beschäftigen.
SARS-CoV-2 und Pandemievorsorge
Während der Coronapandemie haben wir systematisch über 3000 makromolekulare Strukturen aus SARS-CoV-2 evaluiert und die Informationen aus diesen Strukturen zusammengebracht. Wir haben zum Beispiel die Infektion auf molekularer Ebene animiert (https://www.youtube.com/watch?v=aKVXkfR0ygc), und das akkurateste 3D-Modell des Virus erstellt. Nun wollen wir diese Arbeit nutzen, um auch andere potenziell gefährliche Viren – wie zum Beispiel Influenza – zu untersuchen. Mehr Infos unter: www.insidecorona.de.

Pilze
Wir untersuchen Biomoleküle aus Pilzen, die im Wald wachsen. Pilze sind ein relativ unerforschter Organismus in der Strukturbiologie, obwohl sie viele interessante Eigenschaften haben: Sie können sich von sehr ungewöhnlichen Materialien ernähren, binden Schwermetalle aus dem Boden und kommunizieren über weite Strecken. Für diesen Teil unserer Forschung kollaborieren wir unter anderem mit der Abteilung „Mikrobielle Wirkstoffe“ am Helmholtz Zentrum für Infektionsforschung.
Bessere Modelle für die Kristalle biologischer Makromoleküle
In der Kristallographie gibt der R-Wert Auskunft darüber, wie gut ein Modell mit den experimentellen Daten übereinstimmt. Bei kleineren Molekülen werden regelmäßig R-Werte von 3 % erreicht. Bei biologischen Makromolekülen liegen die R-Werte jedoch in der Regel bei etwa 24%. Dieser hohe Wert bedeuteet, dass unsere aktuellen Modelle makromolekularer Kristallstrukturen unvollständig oder teilweise falsch sein können und/oder die Daten enthalten Fehler, die wir noch nicht berücksichtigen. Diese Mängel behindern insbesondere die Bestimmung schwieriger Strukturen, wie z. B. Membranproteine, für die oft nur Daten mit geringer Auflösung verfügbar sind. IIn Zusammenarbeit mit der Gruppe von Armin Wagner am Diamond Lightsource versuchen wir, diese Probleme zu beleuchten und unsere Molekülmodelle biologischer Kristalle zu verbessern, sodass wir nicht nur Grenzfälle lösen, sondern auch alle bekannten Strukturen verbessern können.
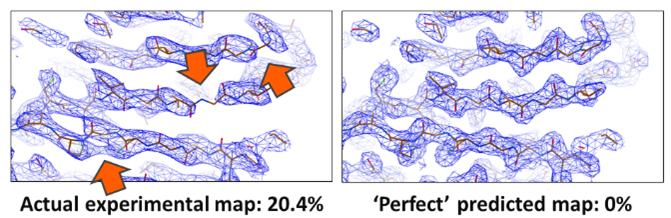
Links: Kristallographische Karte (blaues Gitter); schlechte Phasen dominieren die Karte und es gibt mehrere Probleme (Pfeile), die die Interpretation erschweren. Das Modell (Stäbchen) wird zur Verdeutlichung dargestellt. Rechts: Eine ideale Karte wäre viel einfacher zu interpretieren.
Modellierung großer Komplexe in der Kryo-Elektronenmikroskopie
In der Kryo-EM sind erst seit kurzem Daten mit einer hohen Auflösung verfügbar. Die Kryo-EM kann Strukturen von 100 kDa bis zu mehreren MDa auflösen, benötigt nur eine geringe Menge einer Probe und ist nicht davon abhängig, ob eine Probe kristallisiert werden kann. Da Kryo-EM-Karten einen höheren Informationsgehalt als Röntgendaten haben, sollten sie grundsätzlich den Elektronendichtekarten mit vergleichbarer Auflösung überlegen sein. Derzeit werden jedoch Atommodelle unter Verwendung von Beschränkungen und Parametern, die ursprünglich für die Kristallographie entwickelt wurden, an Rekonstruktionskarten angepasst, was die Antworten, die wir aus den neuen hochauflösenden Daten erhalten können, einschränkt, da die zugrunde liegenden Annahmen nicht immer gerechtfertigt sind. Wir entwickeln Werkzeuge, die direkt auf der Natur der Kryo-EM-Experimente basieren, wie beispielsweise das neuronale Netzwerk Haruspex, um diese Herausforderungen zu bewältigen.
Verbesserung der Datenqualität für die Makromolekülkristallographie
Derzeit führt weniger als 1 % der Messzeit für makromolekulare Strukturen an Synchrotronquellen zu einer veröffentlichten Struktur; ein Großteil der übrigen gesammelten Daten weist vermeidbare Qualitätsprobleme auf – z. B. defekte Kristallproben, fehlerhafte Beugungsexperimente oder eine falsche Interpretation der Bilder durch automatische Datenverarbeitungsverfahren. Viele Daten werden auch als zusätzliche Redundanz gesammelt, da die Datenqualität nur schwer bestimmbar ist. Würden solche Probleme früher und zuverlässiger diagnostiziert, wären die derzeitigen Beamlines wesentlich effizienter. Das Fehlen geeigneter Diagnosemethoden ist eines der größten Hindernisse für die Steigerung der Produktivität von Beamlines für die makromolekulare Kristallographie an Synchrotrons, Freie-Elektronen-Röntgenlasern (XFEL) und Neutronenquellen sowie für die Forschungsqualität in der makromolekularen Kristallographie insgesamt. Jegliche Mängel in den Daten haben unmittelbare negative Auswirkungen auf die resultierende Struktur und damit auf die gewinnbaren biologischen Erkenntnisse. In Zusammenarbeit mit den Mitarbeitern der Beamlines der European X-Ray Free-Electron Laser Facility (European XFEL), der Synchrotronquellen BESSY und ESRF sowie der European Spallation Source ESS entwickeln wir AUSPEX, ein innovatives Diagnosetool, mit dem Beamline-Wissenschaftler und -Nutzer Fehler so früh wie möglich erkennen können, idealerweise noch bevor die eigentliche Datenerfassung beginnt. Wir nutzen diese neuen Werkzeuge auch, um neue Best Practices zu definieren und die Datenverarbeitung zu verbessern. Weitere Informationen hierzu finden Sie unter www.auspex.de.
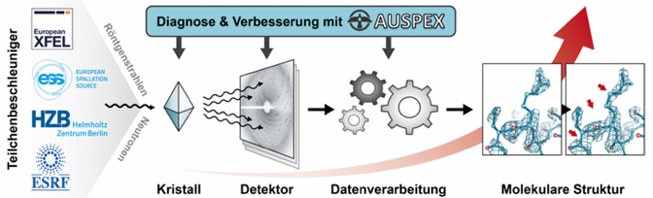
Funktionsweise des AUSPEX-Diagnosetools.